
Professor Sheena Radford
- Position: Astbury Professor of Biophysics and Royal Society Professor
- Areas of expertise: Protein folding/misfolding; amyloid; membrane protein folding; aggregation; biopharmaceuticals
- Email: S.E.Radford@leeds.ac.uk
- Phone: +44(0)113 343 3170
- Location: 10.122a Garstang
- Website: Sheena Radford , Astbury Centre, Leeds | Radford Research Group | X | Bluesky | LinkedIn | Googlescholar | Researchgate | ORCID | White Rose | Open Access REF
Profile
I joined the University of Leeds in 1995 as a Lecturer in the School of Biochemistry and Molecular Biology, progressing to Reader in 1998 and Professor in 2000. In 2009 I became the Deputy Director of the Astbury Centre for Structural Molecular Biology, and its Director in 2012 (until 2021). I became Astbury Professor of Biophysics in 2014 and Royal Society Research Professor in 2021.
Before coming to Leeds, I graduated with a BSc in Biochemistry at the University of Birmingham, completed my PhD in Biochemistry at the University of Cambridge with Professor R.N. Perham, FRS, and held various postdoctoral posts and a Royal Society University Research Fellowship at the Oxford Centre for Molecular Sciences.
I typically supervise 25 PhD students and postdoctoral researchers in my laboratory. In total, more than 160 PhD students and postdocs have progressed successfully from the lab, and have developed their own careers in academic research, industrial posts, and other positions such as technical editing. I am proud of them all.
I have published more than 360 peer-reviewed papers and book chapters and have given more than 475 invited lectures at national and international conferences in countries including the UK, Germany, Denmark, USA, Australia, Japan, Sweden, Ireland, Belgium, Switzerland, Greece, Spain, Italy, France, The Netherlands, Portugal, Croatia, Israel, Austria, Canada, and Thailand, and online.
In the last five years I have served, or am serving on, five major research funding panels and 20 Scientific Advisory Boards for prestigious institutions or companies. I have served on editorial boards for several journals, and am currently Associate Editor of the Journal of Molecular Biology, a Trustee of the Dementia Research Institute, UK and a member of their Council, and the Regional Champion for the Academy of Medial Sciences.
I have been awarded several prizes and awards, including the 1996 Biochemical Society Colworth Medal, the 2005 Royal Society of Chemistry Astra-Zeneca prize in Proteins and Peptides, the 2009 Hites Award from the American Society for Mass Spectrometry (joint with Professor Alison Ashcroft), the Protein Society Carl Branden award in 2013, the 2015 Rita and John Cornforth Award of the Royal Society of Chemistry (also jointly with Professor Alison Ashcroft) and the Biochemistry Society Centenary Award for 2025.
I was elected member of EMBO in 2007, member of Academia Europeae in 2020, Fellow of the Academy of Medical Sciences in 2010, the Royal Society in 2014, and made honorary member of the British Biophysical Society in 2014, a Fellow of the Biophysical Society in 2018, and an Officer of the Most Excellent Order of the British Empire (OBE) in 2020 for services to molecular biology research. In January 2021 I received a Royal Society Research Professorship and was elected a Fellow of the Royal Society of Biology. In 2022 I received the Doctorate honoris causa from the University of Liège, and in 2024 I became an International Member of the National Academy of Sciences (USA).
My research is focused on fundamental structural molecular biology, specifically in the measurement of the conformational dynamics of proteins and the elucidation of the role that these motions play in protein folding and misfolding of both water-soluble and membrane proteins. Using a wide range of biophysical methods and combining these with protein chemistry, molecular biology, chemical biology and structural biology, my research focus over the last 35 years has been the delineation of the mechanisms by which proteins fold or misfold; how dynamic excursions enable proteins to self-associate into amyloid fibrils - the complex macromolecular assemblies associated with some of the deadliest human diseases; and how proteins fold into the bacterial outer membranes of Gram-negative organisms.
Responsibilities
- Astbury Professor of Biophysics
- Royal Society Research Professor
- Group Leader of the Radford Laboratory
Responsibilities
- Astbury Professor of Biophysics
- Royal Society Professor
- Group Leader of the Radford Laboratory
Research interests
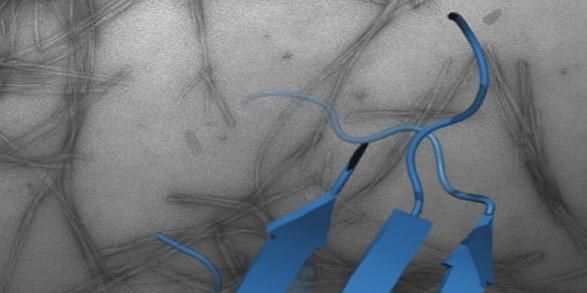
Protein folding, misfolding, aggregation and disease
One of the most fascinating questions in biology is how proteins can fold and assemble into complex, functional entities given just the information provided by the amino acid sequence. A related, equally important facet of the same fundamental question is how protein misfolding can lead to protein aggregation, cellular dysfunction and disease. These issues are the major focus of my research and are tackled using a broad range of techniques including protein chemistry, structural molecular biology, chemical biology, cell biology and biophysical methods.
Current major projects:
- Mechanism(s) of protein misfolding and assembly into amyloid
- Outer Membrane Protein (OMP) folding – The role of chaperones & BAM
- Stabilising proteins of therapeutic interest against aggregation
- Method development (MS, NMR, single molecule, biophysical methods)
Mechanism(s) of protein misfolding and assembly into amyloid

How and why proteins aggregate into amyloid are important fundamental questions that have far-reaching biomedical importance. Focusing on the proteins β2-microglobulin (dialysis related amyloidosis); amylin/IAPP (type II diabetes); α-synuclein (Parkinson’s) and Aβ (Alzheimer’s), work in the group aims to map the structural mechanism(s) of amyloid formation and to develop reagents to control aggregation in vitro and in vivo.
Recent highlights include structure determination of oligomers (NMR, Karamanos et al. eLife (2019) ; Santos et al. JACS (2024)) and fibrils (cryoEM, (with Ranson et al.) (Gallardo et al. Nature Struct. Mol. Biol. (2020)); Wilkinson et al. Nature Comms (2023)), discovery of small molecules that modulate amyloid formation (Cawood et al. JACS (2020), Xu et al. Nature Comms. (2022); Taylor et al. JACS (2025)); and demonstration that early protein-protein interactions in amyloid formation are specific and can be targeted to arrest assembly in vitro and in vivo (Doherty et al. Nature Struct. Mol. Biol. (2020), Ulamec et al. Nature Comms. (2022), Guthertz et al. PNAS (2022); Aubrey et al. PNAS (2024); Dewison et al. PNS (2024)). We have also recently shown that amyloid fibrils structures changes with time (Wilkinson et al. Cell (2023)) and, with Rene Frank and Neil Ranson, have provided the first insights into amyloid in the mouse and human brain of Alzheimer’s patients (Leistner et al. Nat Comms (2023) and Gilbert et al. Nature (2024)).
Outer membrane protein (OMP) – The role of chaperones and BAM

How OMPs fold and assemble into the asymmetric outer membrane (OM) of Gram-negative bacteria is a second research theme in our lab. In a collaborative multidisciplinary team (with David Brockwell, Neil Ranson, Anton Calabrese and Anastasia Zhuravleva) we investigated how OMPs cross the inner-membrane via SecYEG (Crossley et al. EMBO J (2024), In collaboration with Ian Collinson (Bristol)); traverse the periplasm aided by chaperones (Skp/SurA) (Calabrese et al. (2020) Nature Comms) and fold into membranes in vitro and in vivo catalysed by the essential beta-barrel membrane machinery (BAM) (Iadanza et al. (2020) Comms. Biol., Schiffrin et al. (2022) Comms. Biol.)
Building on these insights we are currently exploring the dynamic motions of BAM during catalysis and how this can be harnessed to generate new antibacterial agents against Gram-negative pathogens (White et al. (2021) Nature Comms.). More recently, we have used smFRET and NMR methods to map the binding of client OMPs to their chaperone, SurA (Schiffrin et al. Nat. Comms (2024)) and used cryoEM, smFRET and proteomics to reveal a catalytic cycle of SurA caught in the act of delivering OMPs to BAM (Fenn et al. Nat Comms. (2024)).
Finally, in an on-going collaboration with David Baker (Seattle, USA) and Anastasia Vorobieva (Brussels) we have been exploring how to design transmembrane beta-barrel proteins of different size, along the way teaching us the design principles of this fascinating class of membrane proteins (Vorobieva et al. Science (2021); Berhanu et al. Science (2024)).
Stabilising proteins of therapeutic interest against aggregation

We are also exploiting our knowledge of protein folding/aggregation to practical benefit by screening amyloidogenic proteins, as well as proteins of interest to biopharma, for hotspots that cause aggregation. By coupling aggregation to bacterial growth using a tripartite β-lactamase fusion construct we have discovered small molecules that prevent aggregation of amyloidogenic proteins (Saunders et al. Nature Chem. Biol. (2016)) and are developing the assay further into a deep mutational scanning format.
With David Brockwell and our collaborators and AstraZeneca, we combined the tripartite β-lactamase assay with directed evolution to enhance the resilience of biopharmaceutically-relevant antibodies to aggregation (Ebo et al. (2020) Nature Comms.). In a long-standing collaboration with David Brockwell and Nik Kapur (Mechanical Engineering, Leeds), we are examining how flow fields enhance, or cause, aggregation by flow-induced protein deformations (Dobson et al. PNAS (2017), Willis et al., Eng. Rep. (2020), Willis et al. J. Pharm. Sci. (2024)) and are using the new knowledge to define new ‘rules’ of the design of protein sequences better able to resist biomanufacturing towards making better and cheaper biopharmaceuticals (Willis et al. Mol Pharmaceutics (2025)).
Method development (MS, NMR, single molecule, biophysical methods)

Major developments in methods and instrumentation have played a key role in increasing in our understanding of protein folding and aggregation. Future developments in these fields will also require innovative approaches that cross the boundaries between disciplines. We have been involved in many exciting collaborations to fulfil this aim. With Professors Alison Ashcroft (now emeritus) and Frank Sobott we have developed and expanded our arsenal of methods to interrogate protein folding, protein-protein interactions, and protein complexes, including MS methods (HDX-MS and fast photochemical oxidation of proteins (FPOP-MS)) (Cornwell et al. Analyt. Chem., (2019), Cornwell et al. J. Am. Soc. MS. (2021)); ion mobility MS to determine the effect of ligand binding on amyloidogenic monomers and oligomers (Byrd et al. J. Am. Soc. MS.) (2023) and, with Andrew Wilson (Chemistry), we have developed rapid crosslinking to map protein-chaperone interactions (Horne et al. Angewandte Chemie, (2018) Calabrese et al. Nature Comms (2020)).
Developments in NMR methods remain a mainstay of our laboratory activities (Karamanos et al. Frontiers Neurosci. (2022)). In collaboration with David Brockwell and George Heath we are involved in exciting developments in the use of the AFM for measurements of protein unfolding and protein binding (Ulamec et al. Nature Comms (2021)) and with Paolo Actis (Mechanical Engineering) in the development of nanopores for the manipulation and identification of protein assemblies (Chau et al., Nanoletters (2020), Chau et al. ACS Nano (2022), Chalmers et al. (2024) Nature Comms).
More information about all these projects can be found on the websites of our collaborators’ Astbury and University of Leeds web pages.
For further details about my laboratory, the people involved, molecular images and available opportunities, please visit The Radford Research Group website.
Postdoctoral researchers
Dr Eulashini Chuntharpursat-Bon
Dr Maddie Gilbert
Lectures, seminars and webinars
Discover more about my research journey in this below webinar from the Biochemistry Society (from 9min, 18 seconds). You can also watch and listen to some of my other lectures and webinars on the links below.
How does an IDP aggregate into amyloid? Insights from structural approaches? (2023)
Biophysics of neurodegenerative diseases (2023)
Biochemistry Society Colworth Medal (2023)
Folding a Protein: Nature’s Origami (2022)
Seeing Amyloid: beautiful structures, toxic mechanisms (2020)
Early Steps in Amyloid Assembly: The Achilles Heel of a Disease Mechanism (2020)
Press and news
Links to some of my news stories can be found below.
Leeds academic Professor Sheena Radford wins prestigious Centenary Award from the Biochemical Society, University of Leeds (2024)
Unlocking the power of nanopores, University of Leeds (2024)
Professor Sheena Radford elected to the National Academy of Sciences, University of Leeds (2024)
Shapeshifting fibers: capturing the transformation of a rogue protein, University of Leeds (2024)
Brief encounters: how fleeting protein interactions shape health, University of Leeds (2021)
Leeds scientist backed by Royal Society to transform our understanding of Alzheimer’s, Parkinson’s and nature of memory, Yorkshire Post (2021)
'I'll never stop dancing' says Leeds biophysicist Professor Sheena Radford, who hopes to dispel stereotypes, Yorkshire Post (2019)
Primary investigator (PI)
Co-investigator (Co-I)
- Action in solution: Embedding new technology and new capability in Biomolecular Interactions in the University of Leeds
- BBSRC ALERT 24 - Concurrent force sensing and fluorescence imaging – Enabling the next generation of single molecule biophysics research across Faculties at Leeds
- Breaking the Barrier: Mapping protein interactions in the bacterial outer membrane as targets for new antimicrobials
- Delivery and clearance of outer membrane proteins to the bacterial outer membrane
- EMBeDs: Ensuring Manufacturability of next-generation Biopharmaceuticals by Design
- High-resolution, in situ structures of the blood-brain barrier and neurovascular disease
- Molecular grammar of SurA-client interactions in the periplasm of gram-negative bacteria
- State of the art electron detection for cyoEM at the Astbury Biostructure Laboratories
- Structures and Probes of Intrinsically Disordered Regions
Qualifications
- BSc, Birmingham, 1984
- PhD, Cambridge, 1987
Professional memberships
- 2024 and 2025, Int'l Member of the NAS (USA) & Biochemistry Society Centenary Award for 2025
- 2020, Member of the Academia Europaea, Hon Doctorate, Liège, 2022
- 2021, RS Research Professorship and Fellow of the RS of Biology
- 2020, OBE and Member of the Academia Europaea, 2020
- 2019, Honorary Fellow, St John’s College, Cambridge
- 2017, Fellow of the Biophysical Society for leadership in protein biophysics
- 2014, FRS and Hon Member British Biophysical Society
- 2010, Fellow of Academy of Medical Sciences
- 2007, Fellow of EMBO (European Molecular Biology Organization)
- 2003, Fellow of the Royal Society of Chemistry; also Member of the Biochemical Society and the Protein Society
Student education
I have taught Biochemistry at both undergraduate and postgraduate levels at the University of Leeds since 1995, been an active tutor and supervisor of students at all levels, and, until fairly recently, taught basic mechanisms and concepts in protein folding, and contributed to Advanced Topic Units for final year students. I have been involved with our laboratory’s hosting of students for their final year research projects (Biochemistry and Biological Sciences) for both BSc and MBiol schemes.
Watch the below video to hear how I’m supporting people to navigate a career in science.
Research groups and institutes
- Structural Biology
- Protein structure, stability and dynamics
- Infectious diseases
Current postgraduate researchers
- Chloe Ross-Benham
- Taya M Bosson
- Jack Connor
- Harry Budds
- Luke Johnson
- Rebecca Male
- Thomas Mitchell
- Michael Woodmancy